
Our research
Current focuses in our research
- Epigenetics functions of pRB
- Metabolic functions of pRB
- In vitro cancer stem cell models
An introduction to the RB tumor suppressor gene
A quarter century ago, the identification of the human retinoblastoma gene (RB) loci proved Knudson’s “two-hit theory” that tumor suppressor genes exist. Since then, numerous works delineated crucial roles for the RB protein (pRB)-E2Fs transcription factor complex in G1-S phase transition. In addition, discovering the relationship between pRB and tissue-specific transcription factors enabled a better understanding of how cell cycle exit and terminal differentiation are coupled. Recent works provoked many exciting twists in views on pRB functions during cancer initiation and progression beyond its previously well-appreciated roles. Various mitogenic and cytostatic cellular signals appeared to modulate pRB functions and thus affect a wide variety of effector molecules. In addition, genetic studies in mice as well as other creatures incessantly force us to revise our views on pRB functions for various biological events. This review will focus particularly on the roles of pRB in regulating intracellular signaling, cell metabolism, chromatin function, stem cells and cancer stem cells.
Introduction
RB mutation is found prevalently in retinoblastomas, osteosarcomas and small cell lung carcinomas, and such spectrum of tumors is reproducible in Rb-deficient mice.(1) However, in majority of cancers including prostate, breast, bladder, esophageal, hepatic cancers, glioma or chronic myelogenous leukemia, inactivation of pRB functions caused by either mutation, gene deletion, promoter methylation, deregulated phosphorylation or decreased protein level usually occurs during cancer progression.(2) Some evidence proposes that pRB is even “required“ for tumor initiation in these cancers owing to its anti-apoptotic function or cooperation with Ras-transformation.(3,4) Despite having redundant functions for controlling G1-S transition, other “pocket protein” family members p107 and p130 are rarely mutated in cancers. These findings as well as other discoveries (discussed later) suggest that pRB may possess many more multi-faceted and unique functions than thought previously. Compared with its smaller protein abundance (in empirical terms), pRB appears to possess “too many” functions, and some of them cause both positive and negative effects on the same biological event. This implies that a particular pRB function (or an effect of pRB inactivation) is selected depending on the cellular context or cancer stage.
1. Upstream signals and downstream effectors of pRB
Various mitogenic signals (e.g., receptor tyrosine kinases/Ras, Akt, NF-kB, Shh, Hippo, Wnt, Myc, Jak/STAT) merge more or less on upregulation of D-type cyclins,initiating pRB phosphorylation executed sequentially by cyclinD-CDK4/6 and cyclinE-CDK2 (Figure. 1). This prevents pRB from suppressing E2F function to transactivate the targeted genes. In addition, in response to DNA damage, ATM and Chk1/2 directly phosphorylate pRB.(5) AMP-activated kinase (AMPK) also directly phosphorylates pRB; this contributes to energy control favoring neural progenitor cell growth.(6) Further, pRb phosphorylation induced by AMPK leads to E2F1-dependent cell death that occurs in inner ear cells when they sense mitochondrial defect (discussed later).(7) Oxidative stress resets pRB phosphorylation via protein phosphatase 2A (PP2A).(8) As well documented, the CDK inhibitors (CDKIs) suppress pRB phosphorylation from genetic upstream by attenuating the catalytic activity of cyclin-CDKs. However, clinical outcome of CDKI inactivation is not always equivalent to that of pRB inactivation.(9) pRB inactivation upregulates p16Ink4a by elevating Ras activity and its tumor suppressor role is taken over by p130.(10) This as well as previously described findings indicate that the genetic interaction between CDKIs and pRB is not linear. Furthermore, in addition to phosphorylation, many other types of post-translational modification regulate pRB activity. For instance, pRB is acetylated by p300/CBP, PCAF and Tip60, deacetylated by Sirtuin1 (SIRT1), and methylated by Set7/9 and SMYD.(11,2) These modifications may alter the susceptibility of pRB to undergo CDK-dependent phospholylation or its binding affinity to other partners. Further, pRB is sumoylated, and catabolized following MDM2-mediated ubiquitination or caspase 3 and 8-mediated cleavage at the C-terminus (Figure. 2).(12-14) Mouse models demonstrated that E2Fs are crucial downstream mediators of the tumor suppressor function of pRB. The continuous advancement in our understanding of the E2F functions increases the number of possible functions that pRB may possess (discussed later). Some mutant forms of pRB, however, are defective in E2F binding and transcriptional repression, although the protein partially retain its tumor suppressor activity.(15) This fact led researchers to focus on different downstream effectors. pRB directly binds to Skp2, which allows APC-Cdh1 to ubiquitinate Skp2. Thereby RB loss allows SCFSkp2 E3 ligase complex to bind to and then to ubiquitinate phosphorylated p27Kip1. This nexus appears to be crucial in carcinogenesis since Rb+/-; Skp2-/- mice are completely free from tumor.(16) In addition, EID1 and KDM5A/Jarid1a/RBP2 are recognized as molecules likely to be involved in E2F-independent function of pRB (discussed later).(17-19) pRB family can interact with many enzymes that remodel histones to generate repressive chromatin (Figure. 2 and will be discussed later).(20)
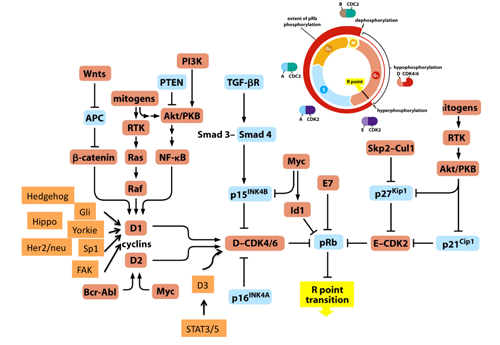
Figure 1. Most of mitogenic signals merge on the transcriptional regulation of D-type cyclins thus target pRB function.
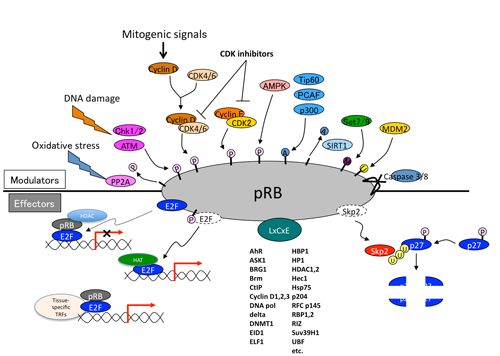
Figure 2. Upstream and downstream (effector) molecules of pRB.
2. Function of pRB in intracellular signaling
(1) Ras signal
Ras activity fluctuates in a cell cycle-dependent manner.(21) Epistasis studies in Caenorhabditis elegans proposed that class B synMuv genes including Rb (lin-35) can control Ras (let-60)-mediated vulval development even from genetic upstream.(22) (Figure 3) Indeed, SV40 large T antigen-mediated pRb inactivation or loss of Rb induced elevated Ras activity in mammalian cells.(23,24) The genetic interaction of Rb and Ras has been analyzed extensively in mouse embryos simultaneously lacking Rb and one of ras isoforms.(25) N- or K-ras deletion significantly prolonged life span of the Rb-null embryos. Their differentiation defects in different organs including muscle cells and erythrocytes were significantly rescued, however aberrant cell proliferation and cell death persisted (results for erythrocytes are unpublished).(26,27) N- or K-ras deletion in Rb-deficient pituitary tumorigenesis impaired tumor invasion with concomitant increase in differentiation degree without affecting tumor incidence.(28,27) These findings implicate that in addition to the previously appreciated pathway in which Ras is upstream of RB, Ras functions also downstream of RB in differentiation control and tumor progression (Figure. 4). Contrary to pituitary, N-ras deletion converts Rb-deficient calcitonin-producing cell (C cell) adenoma or low grade adenocarcinoma to highly metastatic adenocarcinoma.(28) The mechanism of this twist was explained in our later study as following: Rb-deficient C cell adenoma cells are sensitive to DNA damage response induced by mildly elevated N-Ras activity (at most 10-fold elevation in activity compared with that of Rb+ cells, whereas Ras with oncogenic mutation exhibits an approximately 60-fold increase in activity). Subsequently, Rb-deficient C cells undergo “paradoxical” cellular senescence with the aid of p16Ink4a and p130, which protects them from further malignant progression.(10) Consistent with this explanation, Rb-heterozygous mice simultaneously lacking any of Ink4a, Arf or Suv39h1 (senescence-inducing genes) alleles directly developed highly malignant C cell tumors and at an earlier age (Figure 5).(10) This further provided an explanation why RB mutation is a relatively infrequent event during tumor initiation; RB loss-induced carcinogenesis can be antagonized by cellular senescence in some type cells. In a progenitor for human retinoblastoma, RB loss-induced carcinogenesis may be antagonized by p53-dependent apoptosis.(29) However, retinoblastoma progression could be antagonized also by senescence since human RB-null retinomas express elevated p16INK4a and p130 expression, and lose such marks during progression presumably owing to chromosomal instability (CIN) (discussed later).(30) To determine the mechanism that enables pRB to cease Ras activation status, pRb transcriptional targets were determined in Rb-deficient N-ras-/- mouse C cell tumor cells in which proliferation was not affected by the presence of pRb. This enabled the cell cycle-independent function of pRB to be determined. The study detected many genes involved in protein farnesylation and geranylgeranylation (isoprenylation); these post-translational modifications are essential for Ras to be matured and activated. In addition, the study discovered that these genes are dually innervated by E2Fs and sterol regulatory element-binding protein (SREBP) transcription factors. SREBPs are also regulated by E2Fs. In consistent, the same study demonstrated that enhancement of pRb activity delays the trafficking of cytosolic N-Ras to Golgi for which isoprenylation is essential.(10) Not only Ras but also many other small GTPases and CENP-E and F those with CAAX motifs are possibly and RhoA activity was actually regulated by pRB. (28)

Figure 3. The Rb-Ras genetic interaction in C.elegans.
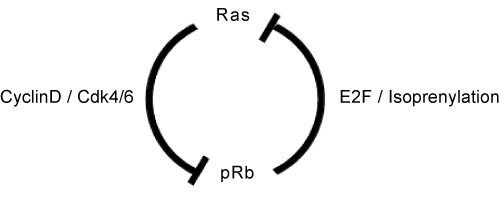
Figure 4. The Rb-Ras genetic interaction in mammalian cells.
(2) Other signals
A study from another group has demonstrated that AKTSer473 phosphorylation is specifically upregulated in cells lacking all RB family members.(31) Although the exact mechanism is still unclear, our own preliminary study demonstrated that the kinetics in which acute pRb inactivation activates AKT seems to be different from that in which pRb inactivation increases GTP-loaded Ras (Kitajima, unpublished data). In addition, elevated Ras activity reportedly prefers to induce phosphorylation at AKTSer308 rather than AKTSer473 via PI3K pathway. Thus, mTORC2 function as well as Ras should be analyzed to discover more on this interaction. Of note, RB and TSC2 (downstream of PI3K/AKT signal) are in a synthetic lethal relationship.(32) The genetic interaction between RB and PI3K/AKT/mTOR signal has just begun to be understood. Myc transcription factors are perhaps one of the most well-recognized pRB transcriptional targets. N-myc gene amplifications are found in retinoblastoma cases free from RB mutation, suggesting that Myc functions may be considerably overlapped with the signals induced by pRB inactivation.(33) In addition, pRB may modulate intracellular signaling by regulating extracellular signaling molecules including VEGF, FGFR, bFGF, matrix metalloproteinases, Interleukin-8, hypoxia-responsive gene products and Cox-2; the machineries may involve E2Fs, Id2, Oct-1, HIF-1 or others.(34,35)
3. Function of pRB in cell metabolism
(1) Metabolic pathways
The aforementioned genetic interaction between RB and Ras mined a new genetic interaction between RB and SREBPs; this further allocated a new role to pRB in lipid metabolism, since SREBPs are master regulators of lipogenic and steroidogenic genes.(10,36) Indeed, in our initial study, pRb appeared to target many of genes coding enzymes that participate in fatty acid and cholesterol biosynthesis. In their promoter, these genes possess either sterol regulatory elements (SREs) or E2F-binding consensus sequences or both (Fig. 4).(10) Recently, a new regulator of SREBP has emerged. Mutated p53 directly binds to SREBP-2 and enhances its transactivation potential, thus contributing to the invasive morphology of breast cancer cells in 3D culture probably owing to enhanced geranylgeranylation.(37) SREBPs transactivate most of genes implicated in the mevalonate (MVA) pathway that governs farnesylation, geranylgeranylation and cholesterol synthesis. This report, in addition to our own study, tightly linked two important tumor suppressors -pRB and p53- to the MVA pathway. Another regulator of SREBP is the PI3K/AKT signaling pathway. An activated AKT signal regulates the SCAP-mediated processing of SREBP precursors, and also attenuates ubiquitination of mature (nuclear) SREBP by inhibiting GSK3 function to phosphorylate mature SREBPs.(38) A recent study demonstrated that Lipin1, which is a substrate for mTORC1 kinase activity, eliminates mature SREBP from the nucleus.(39) Lipin1 could also link SREBP and p53.(40) One of SREBP targets, fatty acid synthase (FASN), has also been identified to be a pRB transcriptional target.(10) This product utilizes NADPH provided by glycolytic pathway and fuels carbon sources into MVA pathway. Elevated FASN level during tumor progression may well explain “lipidogenic phenotype” in cancer cells.(41) This in conjugation with ‘aerobic glycolysis (Warburg’s effect)’ consists two major metabolic perturbations featured in cancer cells. Current understanding is that these machineries synergize to efficiently produce and utilize NADPH for the synthesis of macromolecules including lipids and nucleotides. This occurs while avoiding ROS-producing oxidative phosphorylation (OXPHOS) in mitochondria and while preventing ATP production (Figure 5).(42) In addition, increased cellular cholesterol may suppress OXPHOS by altering the components of mitochondria membrane.(42) NADPH is also required for the synthesis of glutathione, an antioxidant. It is of note that wild type p53 controls glycolysis in a bipolar manner for example by upregulating hexokinase 2 which promotes glycolysis and TIGAR which suppresses glycolysis. In total, mutated p53 is thought to shift cell metabolism to glycolytic (Figure 5). Not only at sequential regulation of SREBP expression and maturation, astonishing level cooperation in regulating cancer cell metabolism is becoming evident between pRB and p53 (Figure 5). AMPKa2 was identified to be an PI3K-sensitive gene group among E2F targets.(43) This molecule functions as a subunit of a system that senses the cellular level of AMP and antagonizes many of metabolic perturbations in cancer cells mostly driven by mTORC1, TCS2 or SREBPs. Therefore, together with the phopshorylation of pRB by AMPK, these findings suggest a mutually suppressive genetic interaction between pRB and AMPK.(6,7) AMPK also functions downstream of another tumor suppressor LKB1.(44) Metformin, an AMPK agonist, was suggested to lower the cancer risk in individuals administered the drug.(45) Although E2F-AMPK genetic interaction was initially perceived in the context of apoptosis control, together with our knowledge on pRB genetic interaction with SREBP, Ras, AKT, Myc, p53, Oct-1 and HIF-1, this discovery will further our understanding of pRB functions in cancer cell metabolism.
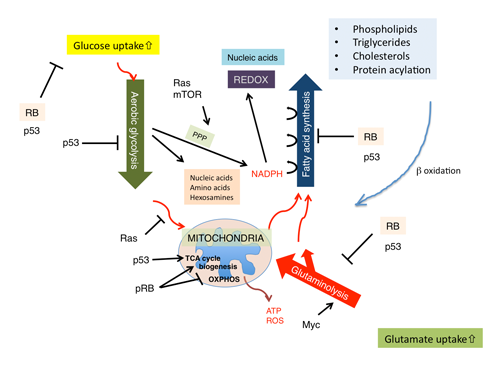
Figure 5. The current model of pRB functions in controlling cell metabolism.
(2) Mitochondrial function
pRB controls mitochondrial biogenesis and function in dissimilar ways. For instance, pRB sustains mitochondrial biogenesis under particular stress conditions. Rb loss induces differentiation defects in cell cycle-exiting erythrocytes and myotube-forming muscle cells presumably by reducing the mitochondrial copy number.(46,47) The latter defect was associated with features of authophagy/mitophagy, and rescued by shifting cells to glycolytic status. This further prompted us to hypothesize that, without glycolytic shift, pRB inactivation may induce shortage of energy to support ATP-consuming differentiation processes (i.e., hemoglobin or myogenic protein synthesis) or even tumor growth. The mechanism linking pRB inactivation to mitophagy may involve a genetic interaction between E2F and Bnip3. (48) pRB also controls transcription of genes involved in mitochondrial functions. This also could overlap with the mechanism by which pRB controls mitochondrial biogenesis. pRB probably through its functional interaction with KDM5A is likely to upregulate number of genes encoding mitochondrial proteins in human monocytic cells.(49) Another study conducted in erythrocytes suggested that pRB affects mitochondrial biogenesis through regulating NRF1, NRF2a, PPARg or PGC-1b in a slightly more complicated manner.(46) A recent twist in this field is that the pRB-E2F-1 complex appeared to suppress genes implicated in OXPHOS.(50) Currently, bifurcated roles of pRB in controlling mitochondrial biogenesis and OXPHOS are unexplained, but this is an intriguing conundrum to solve (Figure 5). In addition, pRB seems to participate in the regulation of cellular reactive oxygen species (ROS) levels, and undergoes feedback from ROS through CDKIs, PP2A, SIRT1 or seladin.(51) Various metabolic pathways controlled by pRB may counterbalance each other in order to keep homeostatic control of cellular metabolism.
4. Function of pRB in chromatin functions
(1) CIN and DNA damage
Increased CIN is one of the representative events seen during progression of cancers. pRB inactivation undermines genome stability through various mechanisms that are E2F-dependent (e.g., via Mad2, cyclin E-driven hyper-replication, nucleotide deficiency, etc) or independent (e.g., via pRB complex with cohesin and condensin II, etc).(52,53) As discussed above, CIN can contribute to retinoblastoma progression. Mad2-induced CIN provides causality to not only tumorigenesis but also to tumor relapse.(54) In addition to such machineries, pRB inactivation can induce DNA damage response and following senescence due to the mildly elevated Ras activity.(10) Because of such diverged roles of pRB in genome stability, the nature of signals generated by DNA damage response induced by pRB inactivation would be different from those induced by other stimuli for example oncogenic Ras or Raf. We are currently investigating how pRb status modulates DNA damage response driven by ATM.
(2) Epigenetic control
A recent study reported that epigenetic changes rather than CIN induced by RB loss contributes more strongly to the retinoblastoma development. Among the genes that are epigenetically regulated by pRB, spleen tyrosine kinase (SYK) appears to play the most critical role.(55) The exact role and mechanism of pRB in epigenetic control is still unclear. Many chromatin modifiers with LxCxE motif including DNMT1 (DNA methyltransferase), Suv39H1, Suv4-20H1 (methyltransferase), HP1 (histone H3me3-binding protein), Brm1, BRG1 (ATP-dependent helicases), HDACs (histone deacetylases) and many of histone demethylases including KDM5A bind to pRB.(20) In addition, new insights on the “metabolic reprogramming” caused in part by pRB and p53 double inactivation has led us to investigate its indirect but noteworthy impact on the chemical modification of chromatin-modifying enzymes by altering the cellular levels of metabolites such as nicotinamide adenine dinucleotide+ (NAD+), flavin adenine dinucleotide (FAD) or a-ketoglutarate.
5. Function of pRB in cell fate decision
The role of pRB in terminal differentiation has been well documented by its ability of binding to or functioning with lineage-specific transcription factors including MyoD, C/EBPa, GRa, GATA-1, PU1, CBFA-1, Pdx1, Runx2 and NF-Il6. In addition, pRB suppresses Id2, KDM5A and EID1, which disturb differentiation. pRB has a crucial role in cell fate decision. For instance, pRB status determines whether p53-mutated osteosarcoma cells accept osteogenic or fat fate.(56) This used to be explained by the functional interaction of pRB with RUNX2 and PPARg. Nevertheless, the impact of “metabolic reprogramming” on differentiation program that stems from the synergistic p53 and pRB inactivation is also a possible explanation. Further, the pRB-p53 alliance appears to determine tumor subtypes through an unknown mechanism. The destruction of this mutually-aiding partnership appears to shift breast and lung cancers to more primitive or epithelial-mesenchymal transition (EMT)-like types.(57,58)
6. Function of pRB in stem cells and cancer stem cells
(1) Tissue stem cells
The intrinsic role of pRB in tissue stem cells has long been debated because studies on the extrinsic role of pRB in hematopoietic stem cells (HSCs) for fetal-maternal nutrient exchange through placenta or bone marrow niche-hematopoietic stem cell adhesion had previously made prevailing impact on the field.(59,60) However, recently, the intrinsic role of pRB in HSCs for oxidative stress response or mitochondrial biogenesis is also appreciated.(51,46) A surprising twist came from plant studies. Arabidopsis thaliana lacking an RB ortholog showed expansion in root stem cell pool without losing self-renewal capacity; this again stimulated a debate on role of pRB in stem cells.(61,62)
(2) Embryonic stem cells
Embryonic stem (ES) cell biology and induced pluripotent stem (iPS) cell technology indicated a requirement of carcinogenic signals for the induction of genome-wide chromatin remodeling and “stemness”. Compared with adult somatic cells, ES or iPS cells exhibit an extraordinarily short G1 phase. The length of G1 could be one of determinants of ES cell fate decision whether to stay in pluripotent state or to differentiate.(63) A short G1 is at least in part due to pRB hyperphosphorylation and suppression of p53 transcription.(64) Nanog seems to maintain pRB at hyperphosphorylated status via CDK6 or CDC25A. (65) On the other hand, loss of p53 promoted the efficacy of iPS induction.(66) The p53-mSin3a-HDACs transcription suppressive complex present on the Nanog gene promoter may be a cell cycle-independent mechanism of this.(67) While an early-published report denied contribution of pRB depletion to iPS induction, one of later studies has demonstrated caspase 3 and 8-mediated pRB cleavage/inactivation facilitates iPS induction.(66,14) Since, pRB is considered to be one of acute targets of some of the pluripotent core factors, knockdown of RB may not show any more additive effect over acute gain of function of these factors.(65) ES cells lacking KDM5A fail prematurely to maintain Oct4 or Nanog expression under differentiation-promoting condition.(68) KDM5A may also mediate the ability of pRB to control mitochondrial biogenesis or functions.(49) Compared with somatic cells, ES and iPS cells contain fewer numbers of mitochondria; cells with far fewer numbers of the organelle maintained high pluripotency but had lower teratoma forming activity (which may be relevant to cell proliferation). Contrariwise, cells with a comparatively high number of mitochondria lose pluripotency but gain higher proliferation activity.(69) It would be intriguing to address whether the status of pRB affects mitochondria biogenesis and pluripotency in ES cells or in cancer stem cells.
(3) Cancer stem cells
Previous studies have detected similarities between poorly differentiated cancer cells and ES cells.(70) Core pluripotent genes, polycomb genes and Myc target genes may play pivotal roles in these shared features.(71) The simultaneous disruption of pRb function and contact inhibition of cells allowed mouse fibroblasts to form a 3D aggregate expressing a variety of core pluripotent genes, and cells derived from such aggregate were also shown to form teratoma-like tumors in immunodeficient mice(72). We developed a similar experimental model, and subsequently identified additional genetic changes that are in fact required for cancer stem cell-like behaviors in Rb-deficient cells (Kitajima et al., unpublished data). Detection of core pluripotent gene transcripts in cancers is often quoted to be a circumstantial evidence of “cancer stemness”, however, its significance is totally unclear. Overexpression of Nanog transforms NIH3T3 fibroblasts and induces cancer stem-like cells from astrocytes.(73,74) Sox2 (as an esophageal “oncogene”) and Nanog have been implicated in the anchorage-independent growth of tumor cells.(75,76) Indeed, phenomenally, anchorage-independent growth is hard to distinguish from sphere formation or cancer transplantability to immuno-deficient mice. However, it is also possible that such circumstantial evidence is non-specifically associated with a genome-wide change in chromatin structure in transformed cells. Attenuation of Rb-deficient pituitary cancers by deletion of the KDM5A loci implies Rb-deficient carcinogenesis may depend on chromatin remodeling.(68) Epigenetic mechanisms are known to affect the causality of human retinoblastoma progression.(55) We observed pRb cooperates with a histone methyltransferase Suv39h1 in suppressing carcinogenesis.(10) pRB inactivation induces cell cycle reentry or tumorigenicity in postmitotic fully differentiated cells with preserving or even reviving their differentiation potential.(77-80) Based on these observations, we may be able to establish new in vivo and in vitro models to address the exact role of pRB in cancer stem cell-like behaviors.
References
- Wikenheiser-Brokamp KA. Retinoblastoma family proteins: insights gained through genetic manipulation of mice. Cellular and molecular life sciences : Cell Mol Life Sci. 2006; 63: 767-80.
- Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008; 8: 671-82.
- Viatour P, Sage J. Newly identified aspects of tumor suppression by RB. Dis Model Mech. 2011; 4: 581-5.
- Williams JP, Stewart T, Li B, et al. The retinoblastoma protein is required for Ras-induced oncogenic transformation. Mol Cell Biol. 2006; 26: 1170-82.
- Inoue Y, Kitagawa M, Taya Y. Phosphorylation of pRB at Ser612 by Chk1/2 leads to a complex between pRB and E2F-1 after DNA damage. Embo J. 2007; 26: 2083-93.
- Dasgupta B, Milbrandt J. AMP-activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development. Dev Cell. 2009; 16: 256-70.
- Raimundo N, Song L, Shutt TE, et al. Mitochondrial Stress Engages E2F1 Apoptotic Signaling to Cause Deafness. Cell. 2012; 148: 716-26.
- Cicchillitti L, Fasanaro P, Biglioli P, et al. Oxidative stress induces protein phosphatase 2A-dependent dephosphorylation of the pocket proteins pRb, p107, and p130. J Biol Chem. 2003; 278: 19509-17.
- Wikenheiser-Brokamp KA. Retinoblastoma regulatory pathway in lung cancer. Curr Mol Med. 2006; 6: 783-93.
- Shamma A, Takegami Y, Miki T, et al. Rb Regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer cell. 2009; 15: 255-69.
- Wong S, Weber JD. Deacetylation of the retinoblastoma tumour suppressor protein by SIRT1. Biochem J. 2007; 407: 451-60.
- Ledl A, Schmidt D, Muller S. Viral oncoproteins E1A and E7 and cellular LxCxE proteins repress SUMO modification of the retinoblastoma tumor suppressor. Oncogene. 2005; 24: 3810-8.
- Uchida C, Miwa S, Kitagawa K, et al. Enhanced Mdm2 activity inhibits pRB function via ubiquitin-dependent degradation. EMBO J. 2005; 24: 160-9.
- Li F, He Z, Shen J, et al. Apoptotic caspases regulate induction of iPSCs from human fibroblasts. Cell stem cell. 2010; 7: 508-20.
- Sellers WR, Novitch BG, Miyake S, et al. Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes Dev. 1998; 12: 95-106.
- Wang H, Bauzon F, Ji P, et al. Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/- mice. Nat Genet. 2010; 42: 83-8.
- Miyake S, Sellers WR, Safran M, et al. Cells degrade a novel inhibitor of differentiation with E1A-like properties upon exiting the cell cycle. Mol Cell Biol. 2000; 20: 8889-902.
- MacLellan WR, Xiao G, Abdellatif M, et al. A novel Rb- and p300-binding protein inhibits transactivation by MyoD. Mol Cell Biol. 2000; 20: 8903-15.
- Benevolenskaya EV, Murray HL, Branton P, et al. Binding of pRB to the PHD protein RBP2 promotes cellular differentiation. Mol Cell. 2005; 18: 623-35.
- Dick FA. Structure-function analysis of the retinoblastoma tumor suppressor protein - is the whole a sum of its parts? Cell Div. 2007; 2: 26.
- Taylor SJ, Shalloway D. Cell cycle-dependent activation of Ras. Curr Biol. 1996; 6: 1621-7.
- Lu X, Horvitz HR. lin-35 and lin-53, two genes that antagonize a C. elegans Ras pathway, encode proteins similar to Rb and its binding protein RbAp48. Cell. 1998; 95: 981-91.
- Raptis L, Brownell HL, Corbley MJ, et al. Cellular ras gene activity is required for full neoplastic transformation by the large tumor antigen of SV40. Cell Growth Differ. 1997; 8: 891-901.
- Lee KY, Ladha MH, McMahon C, et al. The retinoblastoma protein is linked to the activation of Ras. Mol Cell Biol. 1999; 19: 7724-32.
- Takahashi C, Ewen ME. Genetic interaction between Rb and N-ras: differentiation control and metastasis. Cancer Res. 2006; 66: 9345-8.
- Takahashi C, Bronson RT, Socolovsky M, et al. Rb and N-ras function together to control differentiation in the mouse. Mol Cell Biol. 2003; 23: 5256-68.
- Takahashi C, Contreras B, Bronson RT, et al. Genetic interaction between Rb and K-ras in the control of differentiation and tumor suppression. Mol Cell Biol. 2004; 24: 10406-15.
- Takahashi C, Contreras B, Iwanaga T, et al. Nras loss induces metastatic conversion of Rb1-deficient neuroendocrine thyroid tumor. Nat Genet. 2006; 38: 118-23.
- Laurie NA, Donovan SL, Shih CS, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006; 444: 61-6.
- Dimaras H, Khetan V, Halliday W, et al. Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008; 17: 1363-72.
- El-Naggar S, Liu Y, Dean DC. Mutation of the Rb1 pathway leads to overexpression of mTor, constitutive phosphorylation of Akt on serine 473, resistance to anoikis, and a block in c-Raf activation. Mol Cell Biol. 2009; 29: 5710-7.
- Li B, Gordon GM, Du CH, et al. Specific killing of Rb mutant cancer cells by inactivating TSC2. Cancer cell. 2010; 17: 469-80.
- Bremner R, Zacksenhaus E. Cyclins, Cdks, E2f, Skp2, and more at the first international RB Tumor Suppressor Meeting. Cancer Res. 2010; 70: 6114-8.
- Gabellini C, Del Bufalo D, Zupi G. Involvement of RB gene family in tumor angiogenesis. Oncogene. 2006; 25: 5326-32.
- Gauthier ML, Berman HK, Miller C, et al. Abrogated response to cellular stress identifies DCIS associated with subsequent tumor events and defines basal-like breast tumors. Cancer cell. 2007; 12: 479-91.
- Jeon TI, Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab. 2012; 23: 65-72.
- Freed-Pastor WA, Mizuno H, Zhao X, et al. Mutant p53 Disrupts Mammary Tissue Architecture via the Mevalonate Pathway. Cell. 2012; 148: 244-58.
- Krycer JR, Sharpe LJ, Luu W, et al. The Akt-SREBP nexus: cell signaling meets lipid metabolism. Trends Endocrinol Metab. 2010; 21: 268-76.
- Peterson TR, Sengupta SS, Harris TE, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011; 146: 408-20.
- Assaily W, Rubinger DA, Wheaton K, et al. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol Cell. 2011; 44: 491-501.
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007; 7: 763-77.
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011; 11: 85-95.
- Hallstrom TC, Mori S, Nevins JR. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer cell. 2008; 13: 11-22.
- Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009; 9: 563-75.
- Libby G, Donnelly LA, Donnan PT, et al. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes care. 2009; 32: 1620-5.
- Sankaran VG, Orkin SH, Walkley CR. Rb intrinsically promotes erythropoiesis by coupling cell cycle exit with mitochondrial biogenesis. Genes Dev. 2008; 22: 463-75.
- Ciavarra G, Zacksenhaus E. Rescue of myogenic defects in Rb-deficient cells by inhibition of autophagy or by hypoxia-induced glycolytic shift. J Cell Biol. 2010; 191: 291-301.
- Tracy K, Dibling BC, Spike BT, et al. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. 2007; 27: 6229-42.
- Lopez-Bigas N, Kisiel TA, Dewaal DC, et al. Genome-wide analysis of the H3K4 histone demethylase RBP2 reveals a transcriptional program controlling differentiation. Mol Cell. 2008; 31: 520-30.
- Blanchet E, Annicotte JS, Lagarrigue S, et al. E2F transcription factor-1 regulates oxidative metabolism. Nat Cell Biol. 2011; 13: 1146-52.
- Macleod KF. The role of the RB tumour suppressor pathway in oxidative stress responses in the haematopoietic system. Nat Rev Cancer. 2008; 8: 769-81.
- Manning AL, Dyson NJ. pRB, a tumor suppressor with a stabilizing presence. Trends Cell Biol. 2011; 21: 433-41.
- Bester AC, Roniger M, Oren YS, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011; 145: 435-46.
- Sotillo R, Schvartzman JM, Socci ND, et al. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 2010; 464: 436-40.
- Zhang J, Benavente CA, McEvoy J, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012; 481: 329-34.
- Calo E, Quintero-Estades JA, Danielian PS, et al. Rb regulates fate choice and lineage commitment in vivo. Nature. 2010; 466: 1110-4.
- Jiang Z, Deng T, Jones R, et al. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J Clin Invest. 2010; 120: 3296-309.
- Calbo J, van Montfort E, Proost N, et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer cell. 2011; 19: 244-56.
- Wu L, de Bruin A, Saavedra HI, et al. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature. 2003; 421: 942-7.
- Walkley CR, Shea JM, Sims NA, et al. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007; 129: 1081-95.
- Ebel C, Mariconti L, Gruissem W. Plant retinoblastoma homologues control nuclear proliferation in the female gametophyte. Nature. 2004; 429: 776-80.
- Wildwater M, Campilho A, Perez-Perez JM, et al. The RETINOBLASTOMA-RELATED gene regulates stem cell maintenance in Arabidopsis roots. Cell. 2005; 123: 1337-49.
- Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008; 9: 115-28.
- Miura T, Luo Y, Khrebtukova I, et al. Monitoring early differentiation events in human embryonic stem cells by massively parallel signature sequencing and expressed sequence tag scan. Stem Cells Dev. 2004; 13: 694-715.
- Conklin JF, Sage J. Keeping an eye on retinoblastoma control of human embryonic stem cells. J Cell Biochem. 2009; 108: 1023-30.
- Hong H, Takahashi K, Ichisaka T, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009; 460: 1132-5.
- Lin T, Chao C, Saito S, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005; 7: 165-71.
- Lin W, Cao J, Liu J, et al. Loss of the retinoblastoma binding protein 2 (RBP2) histone demethylase suppresses tumorigenesis in mice lacking Rb1 or Men1. Proc Natl Acad Sci U S A. 2011; 108: 13379-86.
- Schieke SM, Ma M, Cao L, et al. Mitochondrial metabolism modulates differentiation and teratoma formation capacity in mouse embryonic stem cells. J Biol Chem. 2008; 283: 28506-12.
- Ben-Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008; 40: 499-507.
- Kim J, Woo AJ, Chu J, et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010; 143: 313-24.
- Liu Y, Clem B, Zuba-Surma EK, et al. Mouse fibroblasts lacking RB1 function form spheres and undergo reprogramming to a cancer stem cell phenotype. Cell stem cell. 2009; 4: 336-47.
- Piestun D, Kochupurakkal BS, Jacob-Hirsch J, et al. Nanog transforms NIH3T3 cells and targets cell-type restricted genes. Biochem Biophys Res Commun. 2006; 343: 279-85.
- Moon JH, Kwon S, Jun EK, et al. Nanog-induced dedifferentiation of p53-deficient mouse astrocytes into brain cancer stem-like cells. Biochem Biophys Res Commun. 2011; 412: 175-81.
- Bass AJ, Watanabe H, Mermel CH, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009; 41: 1238-42.
- Jeter CR, Badeaux M, Choy G, et al. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells. 2009; 27: 993-1005
- Sage C, Huang M, Karimi K, et al. Proliferation of functional hair cells in vivo in the absence of the retinoblastoma protein. Science. 2005; 307: 1114-8.
- Mantela J, Jiang Z, Ylikoski J, et al. The retinoblastoma gene pathway regulates the postmitotic state of hair cells of the mouse inner ear. Development. 2005; 132: 2377-88.
- Pajcini KV, Corbel SY, Sage J, et al. Transient inactivation of Rb and ARF yields regenerative cells from postmitotic mammalian muscle. Cell stem C
ell. 2010; 7: 198-213. - Ajioka I, Martins RA, Bayazitov IT, et al. Differentiated horizontal interneurons clonally expand to form metastatic retinoblastoma in mice. Cell. 2007; 131: 378-90